This paper describes joint work with Alexis Battle, Sara Mostafavi, and Daphne Koller. Thanks as well to the GTEx Consortium for data.
In spite of what this blog might imply, I do not spend all my time studying sex and stalking people on Twitter. From nine to five I apply statistics to biological datasets because a) there is an enormous amount of new biological data with exciting implications for how we treat disease and b) there is no way to make sense of it without using a computer. This week we published a paper on which I began work as an undergrad, and on the theory that one should never do work that cannot be explained to the intelligent (and attractive) non-specialist which I imagine you are, I am going to describe it here. This post is a little longer than most of my posts, mostly because it has lots of pictures, but I have faith in you.
We begin with a mystery: cells throughout your body have (essentially) the same DNA, and yet do very different things. How? One sentence recap of ninth grade bio:
Genes (blueprints for proteins) are used to produce RNA (an intermediate molecule) which is used to produce proteins (tiny molecular machines that do most of the work in a cell)
So by altering how much RNA you make from a gene, you can control the amount of protein, and thus the functions of a cell. We call this RNA data “gene expression data”, and recently a large group of scientists known as the GTEx Consortium produced gene expression data for more than a thousand samples from dozens of different human tissues. This data allows you to ask cool questions like “what genes are particularly highly expressed in the liver?” which gives you a hint about which genes are important to the liver’s functions.
Another thing we might expect to differ between tissues is how gene expression levels are correlated with each other. (Genes A and B are positively correlated if, when A is expressed at a high level, so is B.) We say that correlated genes are “co-expressed”, and we care because genes that are co-expressed often work together. So looking at the specific co-expression network for each tissue can tell us something about each tissue accomplishes its function. Below is an example of a co-expression network (source): genes are circles, co-expressed genes are linked by blue lines, and you can see that genes with common functions often cluster together .
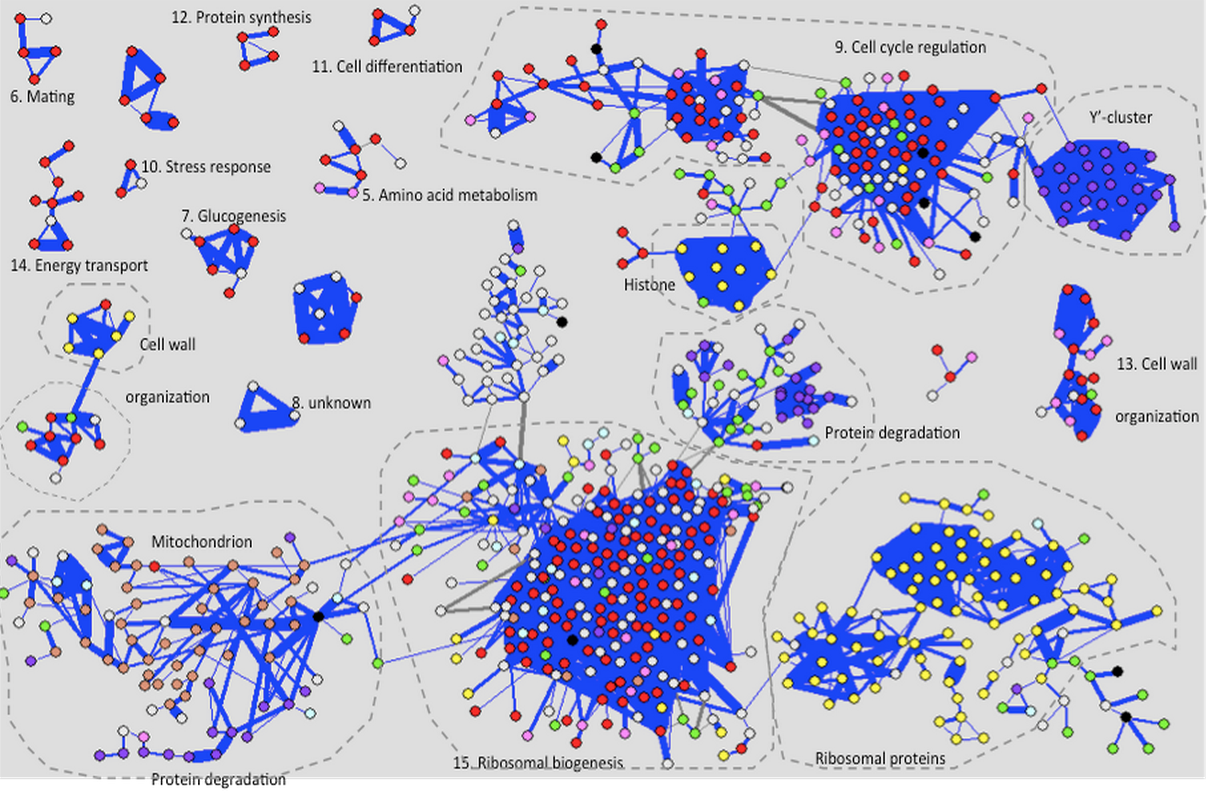
In our paper, we study co-expression networks in 35 tissues throughout the human body, and we do three things:
- We come up with a new method for inferring the networks more accurately. I named this method “GNAT” (Gene Network Analysis Tool), for my boyfriend Nat, because I am a computer scientist and this is the sort of thing we think is romantic.
- We statistically study the networks to find biological principles.
Inferring the networks is hard because you have tens of thousands of genes and you are considering links between all possible pairs of genes. This means that with ten thousand genes you have about fifty million possible links. This raises two issues.
1. The sheer size of the mathematical objects involved can crash your computer or take way too long to deal with. GNAT uses various mathematical tricks to deal with this.
2. You don’t have enough data. For some tissues, you might have samples from only a dozen people, which is not enough to give you very good estimates of fifty million links. GNAT improves the estimates by sharing information across tissues: while the networks in your liver and brain are different, they probably also have some similarities, and so we can use the brain’s network to learn the liver’s more accurately. We make a tree that groups similar tissues together, and we encourage tissues close together in the tree to have similar networks. We show that this increases accuracy. While we use our method on human tissues, you could also use it on any group of datasets related by a tree: different species, evolving cancer cells, or even non-biological correlation datasets. Here is the tree we used:

Having learned the networks, we analyze them to try to find principles that guide how the body works. We look at two types of genes: transcription factors, which are master regulators that control lots of other genes, and genes which are known to have tissue-specific functions (for example, immune-related genes in the blood). If your genes were the Mafia, transcription factors would be the kingpins and tissue-specific genes would be the street-level enforcers (that’s how I thought about it, anyway). We find that the kingpins tend to be connected to the enforcers, and enforcers connected to kingpins are expressed at higher levels. Kingpins tend to lie at the centers of networks, while enforcers tend to lie at the peripheries. All this paints a coherent statistical picture of how tissues accomplish their specific functions: tissue-specific kingpins control and upregulate tissue-specific enforcers.
We also find lots of groups of interconnected genes that may work together. Groups that are highly expressed in a particular tissue often have tissue-specific functions: for example, neural firing in the brain and muscle function in the heart. Groups that persist across tissues tend to have functions common to many types of cells, like cell division.
On the one hand, it is deeply cool to find mathematical evidence of deep biological principles. While working on this paper I went for a walk around Stanford’s Dish and on one particularly steep ascent (photo cred to Shengwu Li)

I could feel my lungs and muscles working and I thought: I have seen the clusters of genes that let my lungs bring me this air and my blood fight its pathogens and my muscles use its oxygen. I’m not a religious person, so moments like this are about as close as I get to spirituality. At the same time, it is hard to verify the links in our networks: because our analysis is based on correlations, we need more targeted biological techniques to firmly establish causality. We are also peering into a deeply alien world about which we have relatively little data. I enjoy working with social science data because I have an intuition for what the confounds and interesting questions are; high-dimensional biological data remains huge and unintuitive to me. Still, it’s pretty cool that we’re kept alive every second by processes so far beyond our understanding -- stare in reverence at your palms, because that skin conceals a million microscopic mysteries.
The last thing we did was make a web tool so other people could use our networks for their own discoveries. The story of making this tool illustrates how research often works for me, so I will tell it because I think it’s useful to not just present final products. On the left is the original version, which I wrote; on the right is the pretty and much faster version, which my coauthors wisely hired a professional web programmer to revamp. (Definitely play with the latter if you’re actually curious.)
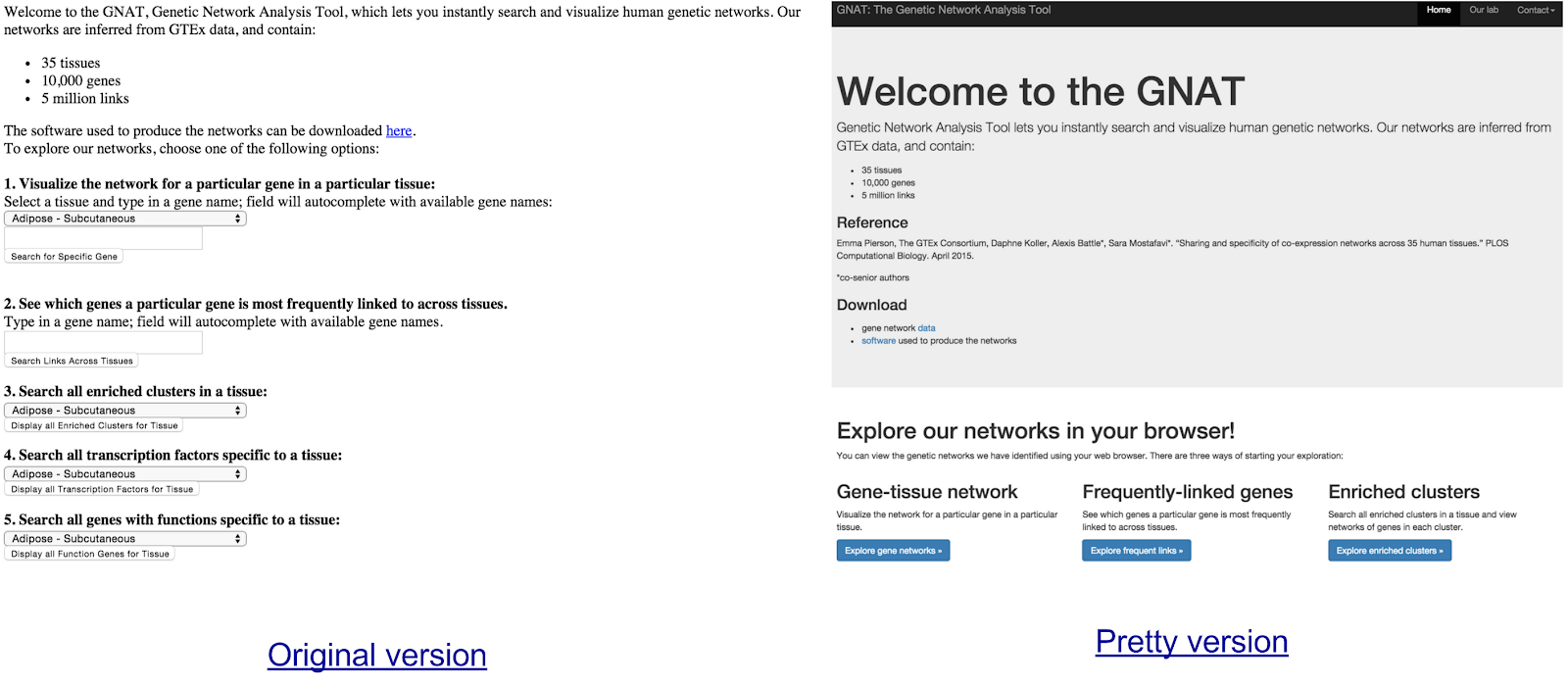
I liked my coauthors’ idea to build a website but had never made one. So I didn’t pursue it for a while, since the website would have to A) process very large genetic networks quickly and B) make complex graphics from them:
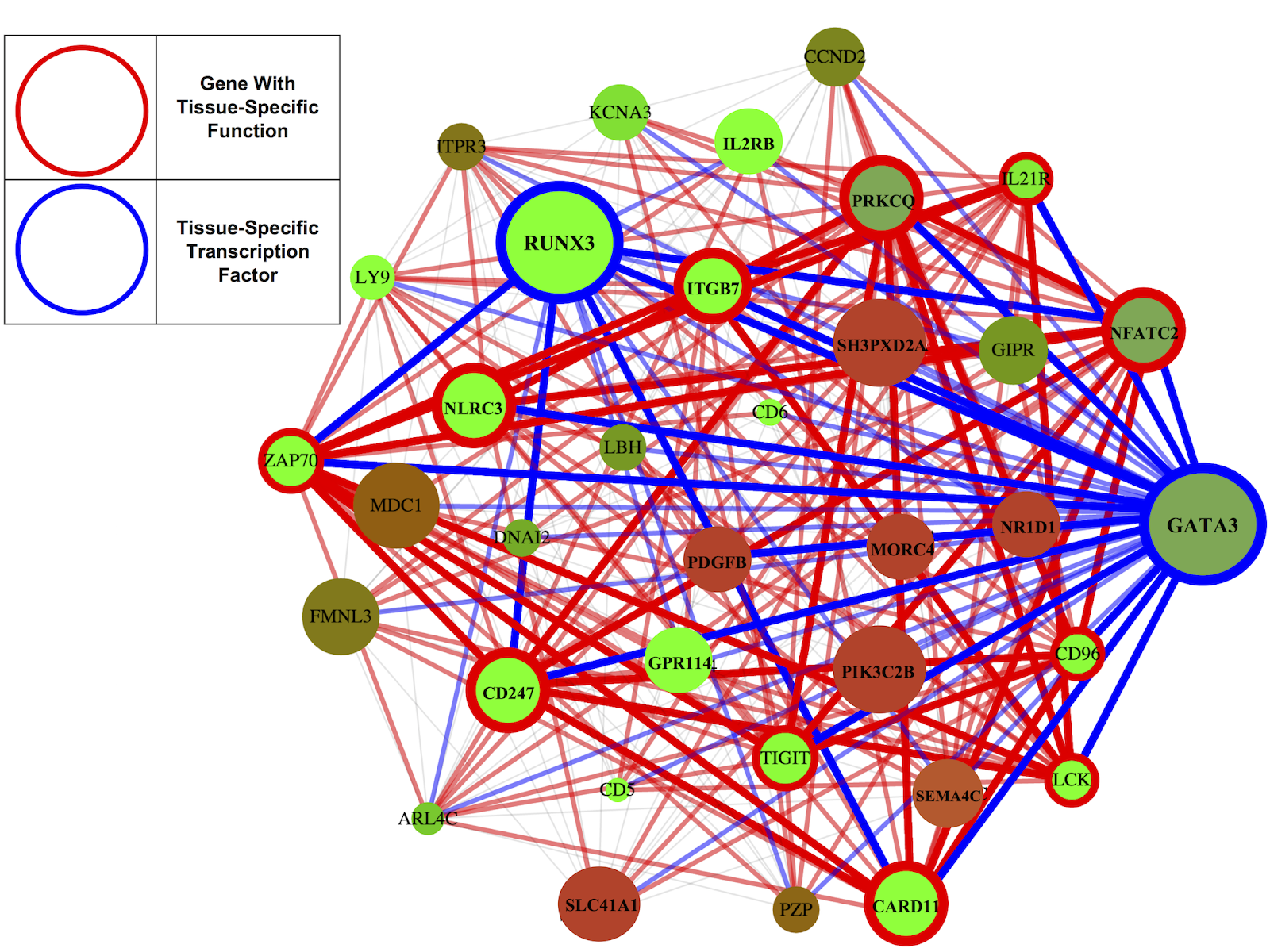
and I didn’t know how to do either. But that summer I was working at Coursera, and one day my boss showed me Flask, which is a tool that allows you to accomplish A. Then I went out to dinner with Sophia Westwood and Sarah Sterman, two computer science master's students, and Sophia showed us a network visualization she had built using a tool called d3. She was visualizing computer programs, not genes, but the similarity was there, and that showed me how to do part B.

The moral of this story, which I have learned repeatedly, is that hanging out with smart people is always useful because it gives you new ideas in unexpected directions. Relatedly, I learned a ton from working on this paper, mostly from my coauthors (two of whom became professors while we were writing it). The contrast in scale with my social science projects is striking: those usually take weeks, and this took years. I am still trying to decide what combination of blog posts, general audience pieces, and academic publications most efficiently gets new information to the people who will benefit from it.
I used this term paper writing tips before applying to my university, must be helpful for newbies! So look on it!
ReplyDeletesmart defrag pro crack
ReplyDeleteradioboss crack
minitool power data recovery crack
n-track studio crack
movavi video converter crack
coolutils total pdf converter crack
easeus mobimover full crack
driver reviver crack
ableton live crack
minitool power data recovery crack
Thank you for sharing this insightful post. Take a moment to visit this profile Spacebar Clicker. The spacebar clicker is excellent for refining speed and accuracy.
ReplyDelete